В том случае, когда величина Kp неизвестна ни при одной из температур, Const в уравнении
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT + Const·T
остается неопределенной постоянной величиной. Проблема определения постоянной интегрирования в вышеприведенном уравнении, минуя экспериментальное определение константы равновесия, привлеклав конце XIX – начале XX века внимание многих физико-химиков. В 1906 году Нернст в своей работе «О вычислении химического равновесия из термических данных» высказал постулат, согласно которому
кривые в координатах ΔrG°(T) – T и ΔrH°(T) – T для любой химической реакции в конденсированных системах вблизи абсолютного нуля асимптотически приближаются друг к другу, т. е. имеют общую касательную при T = 0K:
T → 0lim∂(ΔrG°)∂Tp = T → 0lim∂(ΔrH°)∂Tp – тепловая теорема Нернста.
Из данной теоремы вытекает ряд важных следствий.
1–е следствие из тепловой теоремы Нернста
Вспомним уравнениеГиббса-Гельмгольца
ΔrG° = ΔrH° + T∂(ΔrG°)∂Tp.
Откуда: ∂(ΔrG°)∂Tp = ΔrG° – ΔrH°T .
Перейдем к пределу приT → 0:
T → 0lim∂(ΔrG°)∂Tp = T → 0limΔrG° – ΔrH°T = 0 0 ,
т. е. получаем неопределенность. Для раскрытия неопределенности нужно найти предел отношения производных по температуре от числителя и знаменателя:
т. к. числитель равен нулю,а знаменатель – единице. Отсюда:
T → 0lim∂(ΔrG°)∂Tp = T → 0lim∂(ΔrH°)∂Tp = 0.
Таким образом, в соответствии с первым следствием касательная к кривымΔrG°(T) = f(T) и ΔrH°(T) = f(T) должна быть параллельной оси температур:
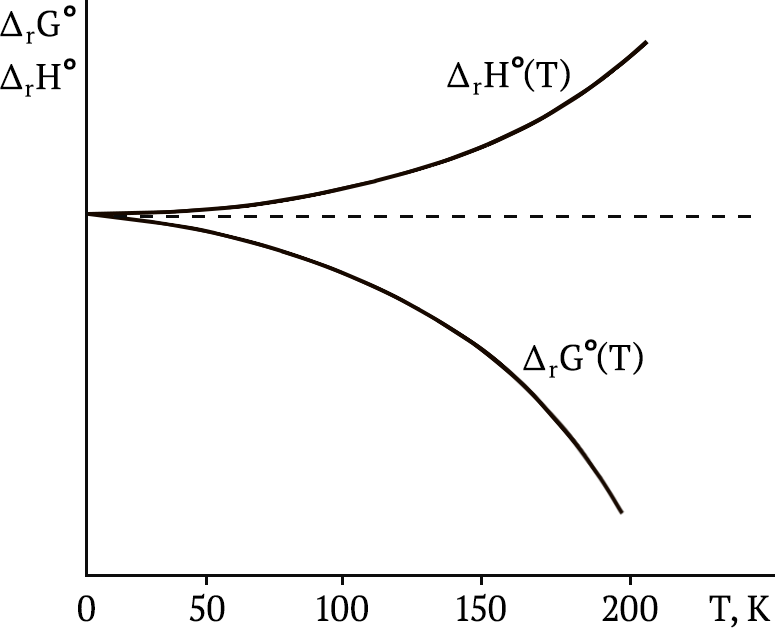
2–е следствие из тепловой теоремы Нернста
Вспомним, что
Cp = ∂H∂Tp или Δ(νCp°) = ∂(ΔrH°)∂Tp,
откуда: T → 0limΔ(νCp°) = 0.
Это значит, что приT → 0 химические реакции в конденсированных системах протекают без изменения теплоемкости системы. При этом, как показывают опыт и квантовая теория, для правильных кристаллов не только
T → 0limΔ(νCp°) = 0 ,
но и теплоемкость каждого из компонентов системы приT → 0 стремится к нулю, т. е.:
T → 0limCv = 0
и T → 0limCp = 0.
3–е следствие из тепловой теоремы Нернста
Вспомним, что
∂G∂Tp = –S или ∂(ΔrG°)∂Tp = –ΔrS°,
откуда: T → 0limΔrS° = 0и T → 0lim S = 0.
4–е следствие из тепловой теоремы Нернста
Комбинируя уравнениеG = H – TS с уравнениями
ΔrH°(T) = ΔrH°(0) + 0 T Δ(νCp°)dT,
ΔrS°(T) = ΔrS°(0) + 0 T Δ(νCp°)TdT,
получаем
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT – TΔrS°(0).
Откуда, с учетом уравнения
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT + Const·T,
следует, что Const = –ΔrS°(0). А поскольку изменение энтропии ΔrS°(0) в процессах между кристаллическими веществами равно нулю, постоянная интегрирования Const также равна нулю. Откуда:
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT.
5–е следствие из тепловой теоремы Нернста
Для реакции
ν1A + ν2B ⇄ ν3C + ν4D,
протекающей в газовой фазе, постоянная Const′ в уравнении
lnKp = 1R ∫ ΔrH°T2dT + Const′ ,
равна алгебраической сумме истинных химических постоянных j:
Const′ = (ν3jC + ν4jD) – (ν1jA + ν2jB) = Δj.
Значения истинных химических постоянных могут быть вычислены по экспериментальным данным зависимости давления насыщенного пара от температуры или методами статистической термодинамики, согласно которым
j = S(0) – Cp(0)R,
гдеS(0) и Cp(0) – абсолютная мольная энтропия и изобарная теплоемкость вещества в газообразном состоянии при T = 0K. Поскольку для реакции в газовой фазе Const′ = Δj, то
lgKp = – ΔrH°(0)RT + 1R 0 T dT T2 0 T Δ(νCp°)dT + Δj.
Таким образом, константу равновесия Kp можно вычислить на основании только термических данных для участников реакции.
Большинство современных методов расчета химического равновесия базируется на третьем начале термодинамики, которое формулируется следующим образом:
энтропия любого индивидуального бездефектного кристаллического вещества (идеального кристалла) при абсолютном нуле равна нулюS(0) = 0 .
Как было сказано ранее, это положение впервые было высказано в виде постулатаПланком в 1912 году .
Итак, третье начало термодинамики не является точным утверждением. Вычисленные на основании калориметрических данных абсолютные значения энтропии носят условный характер. Несмотря на это, третье начало термодинамики имеет большое практическое значение. На его основе разработаны современные методы вычисления стандартных функции Гиббса реакций, а также констант равновесия при различных температурах.
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT + Const·T
остается неопределенной постоянной величиной. Проблема определения постоянной интегрирования в вышеприведенном уравнении, минуя экспериментальное определение константы равновесия, привлекла
Из данной теоремы вытекает ряд важных следствий.
Вспомним уравнение
ΔrG° = ΔrH° + T∂(ΔrG°)∂Tp.
Откуда: ∂(ΔrG°)∂Tp = ΔrG° – ΔrH°T .
Перейдем к пределу при
T → 0lim∂(ΔrG°)∂Tp = T → 0limΔrG° – ΔrH°T = 0 0 ,
т. е. получаем неопределенность. Для раскрытия неопределенности нужно найти предел отношения производных по температуре от числителя и знаменателя:
| = 0, | ||
| T → 0lim∂T∂T |
т. к. числитель равен нулю,
Таким образом, в соответствии с первым следствием касательная к кривым
Вспомним, что
Cp = ∂H∂Tp или Δ(νCp°) = ∂(ΔrH°)∂Tp,
откуда: T → 0limΔ(νCp°) = 0.
Это значит, что при
но и теплоемкость каждого из компонентов системы при
Вспомним, что
∂G∂Tp = –S или ∂(ΔrG°)∂Tp = –ΔrS°,
откуда: T → 0limΔrS° = 0
Комбинируя уравнение
ΔrH°(T) = ΔrH°(0) + 0 T Δ(νCp°)dT,
ΔrS°(T) = ΔrS°(0) + 0 T Δ(νCp°)TdT,
получаем
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT – TΔrS°(0).
Откуда, с учетом уравнения
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT + Const·T,
следует, что Const = –ΔrS°(0). А поскольку изменение энтропии ΔrS°(0) в процессах между кристаллическими веществами равно нулю, постоянная интегрирования Const также равна нулю. Откуда:
ΔrG°(T) = ΔrH°(0) – T 0 T dT T2 0 T Δ(νCp°)dT.
Для реакции
ν1A + ν2B ⇄ ν3C + ν4D,
протекающей в газовой фазе, постоянная Const′ в уравнении
lnKp = 1R ∫ ΔrH°T2dT + Const′ ,
равна алгебраической сумме истинных химических постоянных j:
Const′ = (ν3jC + ν4jD) – (ν1jA + ν2jB) = Δj.
Значения истинных химических постоянных могут быть вычислены по экспериментальным данным зависимости давления насыщенного пара от температуры или методами статистической термодинамики, согласно которым
j = S(0) – Cp(0)R,
где
lgKp = – ΔrH°(0)RT + 1R 0 T dT T2 0 T Δ(νCp°)dT + Δj.
Таким образом, константу равновесия Kp можно вычислить на основании только термических данных для участников реакции.
Большинство современных методов расчета химического равновесия базируется на третьем начале термодинамики, которое формулируется следующим образом:
энтропия любого индивидуального бездефектного кристаллического вещества (идеального кристалла) при абсолютном нуле равна нулю
Как было сказано ранее, это положение впервые было высказано в виде постулата
Итак, третье начало термодинамики не является точным утверждением. Вычисленные на основании калориметрических данных абсолютные значения энтропии носят условный характер. Несмотря на это, третье начало термодинамики имеет большое практическое значение. На его основе разработаны современные методы вычисления стандартных функции Гиббса реакций, а также констант равновесия при различных температурах.